AlphaFold1
[Summarize] AlphaFold1 Review
1. 연구 목적
논문에서는 1차원으로 연결된 아미노산 서열의 3차원 구조를 예측하는 것을 목적으로 합니다. 아래 그림(figure 1.)에서 보듯이 “MKWPEPTF” 순서로 구성된 아미노산들은 a. 그림처럼 1차원으로 표현할 수 있으며, 하나의 아미노산(그림에서 원)은 카복시기, 아미노기, 잔기(residue)로 구성되어 있습니다. 서로 다른 아미노산의 잔기들이 가진 전자기적 특성으로 인해 b. 그림과 같이 접히는 성질을 보입니다.
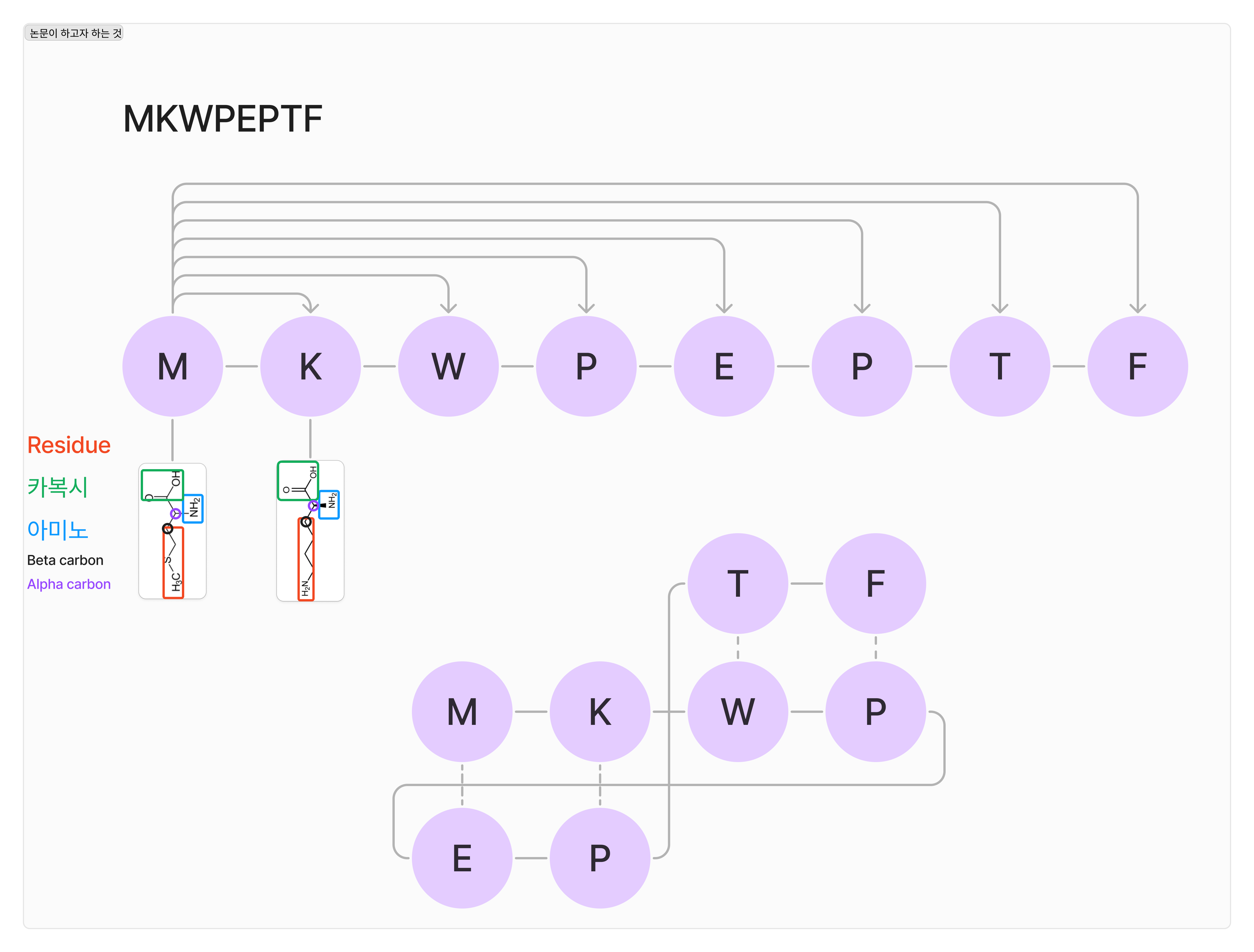
figure 1. 원으로 표현되어 있는 것은 하나의 아미노산이며 이러한 아미노산이 연결되어 있는 단백질은 자연에서 1열 형태(a. 이미지) 보다 3차원 구조로 접혀 있다.
1.1 Why do we need it??
단백질은 체내에서 혹은 자연에서 특정 기능을 수행하는 특징이 있습니다. 이러한 기능은 단백질의 3D 구조를 통해 유추할 수 있습니다. 신약 개발 과정에서는 목적 기능(예: 특정 바이러스의 기능을 억제하도록 설계된 단백질)을 수행하는 단백질을 만들기 위해, 후보 아미노산 서열을 직접 생성하고 이를 관찰하여 원하는 단백질 구조가 형성되었는지 확인합니다. 하지만 이 방법은 오랜 시간과 많은 인력, 비용이 소요되어 더 효율적인 방법이 필요했습니다. 이러한 배경에서 아미노산 서열을 입력하면 단백질의 3차원 구조를 예측하는 시뮬레이터를 개발하고자 하였습니다.
1.2 기존 실험적 방법의 한계
단백질 구조 결정을 위한 기존의 실험적 방법들은 다음과 같은 기술들이 있습니다:
- X-선 결정학 (X-ray Crystallography)
- 극저온 전자현미경 (Cryo-EM)
- 핵자기공명 분광법 (NMR Spectroscopy)
이러한 방법들은 매우 정확한 구조 정보를 제공하지만, 다음과 같은 한계가 있습니다:
- 많은 시간과 비용이 소요됨
- 모든 단백질에 적용할 수 없음 (예: 결정화가 어려운 단백질)
- 특수한 실험 조건이 필요함
- 전문 인력과 고가의 장비가 필요함
이러한 한계로 인해 현재까지 실험적으로 구조가 결정된 단백질은 전체 알려진 단백질 서열의 극히 일부에 불과합니다.
2. 방법
2.1 Input Data
단백질이 접히는 현상은 아미노산의 residue(잔기) 특성으로 부터 발현됩니다. 이러한 근거로 논문에서는 residue의 정보들을 기반으로 단백질의 3차원 구조를 예측합니다.
논문에서는 residue의 정보를 두 가지 방법으로 접근합니다:
- 하나의 residue에서 얻을 수 있는 정보 (1D 정보)
- residue의 쌍에서 얻을 수 있는 정보 (2D 정보)
이 둘 정보를 3차원 정보로 합쳐 사용합니다. 여기서의 3차원은 1. 연구목적에서의 3차원 구조와는 다른 개념입니다. 앞서 설명한 3차원은 물리 세계에서의 3차원으로 가로 세로 높이 좌표에 매핑되는 물체를 이야기하는 것이며, 여기서 설명하는 3차원 정보는 어떠한 정보를 3개의 차원으로 표현하는 데이터를 말하는 것입니다.
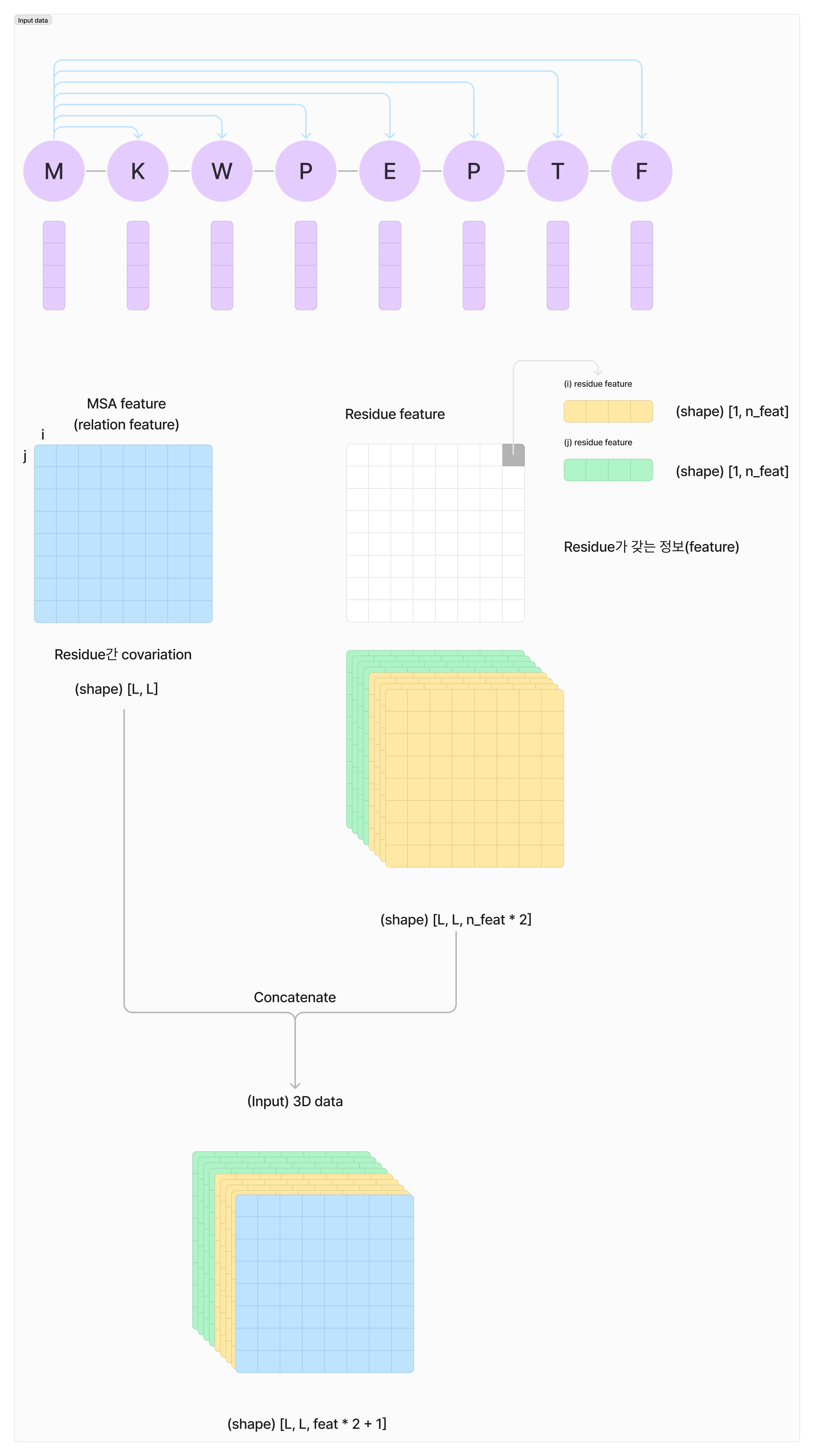
figure 2.
2.1.1 MSA 데이터
앞서 Residue 간의 관계를 나타내는 정보를 MSA(Multiple Sequence Alignment, 다중 서열 정렬)을 통해서 얻어오게 됩니다. MSA는 서열간의 상호작용과 공진화(covariation) 정보를 추출하여 단백질 구조를 결정하는 데 필요한 단서를 제공합니다.
MSA 데이터는 다음과 같은 데이터베이스를 활용합니다:
- Uniclust30 (2017-10)
- PSI-BLAST nr dataset (2017-12-15)
- PDB (2018-03-15)
- CATH (2018-03-16)
2.1.2 MSA의 역할
MSA는 단백질 서열의 진화적 관계를 통해 특정 위치에서의 아미노산 상호작용 정보를 제공합니다. 이를 통해 다음을 파악할 수 있습니다.
- 공진화 정보: 단백질 서열에서 특정 위치의 아미노산들이 서로 상호작용하며 진화적으로 연관되어 있다는 정보
- 예: 한 위치의 변화가 다른 위치의 변화와 동반될 경우, 두 위치가 구조적으로 인접할 가능성이 높음
- 보존된 서열 정보: 진화적으로 보존된 아미노산 위치는 단백질의 핵심 구조적 또는 기능적 역할을 나타냄
AlphaFold에서 2D로 받아오는 MSA 정보
- 서열 간 상화작용 정보를 2D 행렬로 표현:
- 공변이 행렬(corariance Matrix):
- MSA에서 두 위치 간의 상관관계(공진화)를 계산하여, 이들이 구조적으로 인접할 가능성을 나타냄
- 예: 위치 i와 j에서 아미노산 변이가 동시에 발생하면, 이 둘이 구조적으로 상호작용할 가능성이 큼
- 연결 정보(Contact Map):
- 공변이 행렬로부터 두 아미노산 사이의 물리적 근접 가능성을 예측
- 이는 단백질의 3D 구조를 정의하는 데 매우 중요
- 공변이 행렬(corariance Matrix):
2.1.3 MSA pros and cons
pros:
- 진화 정보 제공:
- MSA는 단백질 서열 간의 진화적 관계를 기반으로 구조 예측에 중요한 단서를 제공
- 공진화 정보 활용:
- 공변이 분석을 통해 단백질의 3D 구조를 예측할 수 있는 구조적 단서를 제공
- 보존 영역 식별:
- 기능적으로 중요한 보존된 아미노산 위치를 식별하여 단백질의 기능적 특성을 이해
cons:
- MSA 품질 의존성:
- MSA의 품질이 낮거나 관련 서열 데이터가 부족하면, 구조 예측 정확도가 크게 떨어질 수 있음(새롭게 발견된 아미노산 서열에 대한 구조는??)
- 계산 비용:
- MSA 생성은 대규모 데이터베이스에서 검색하고 정렬하는 데 시간이 많이 소요됨
- 진화적 정보 제한:
- 잘 보존되지 않은 단백질(예: 드문 서열)의 경우 MSA에서 충분한 정보를 얻기 어려움
- 노이즈:
- 서열 정렬 과정에서 잘못된 갭 삽입 등으로 인해 불필요한 노이즈가 추가될 수 있음
2.1.4 데이터 병합
아미노산 서열로 부터 얻을 수 있는 1D 데이터와 2D 데이터를 하나의 데이터로 병합하는 과정을 (figure 2.)에서 설명하고 있다. MSA로 얻은 2D 데이터는 하나의 픽셀(한 칸) i 번째 아미노산과 j 번째 아미노산의 관계를 만들고 있으며 1D 데이터에는 각각의 아미노산의 정보들을 뒤에 쌓는 방법으로 하나의 데이터로 병합하고 있다.
1D 데이터는 하나의 아미노산(residue)에서 표현하는 정보를 담아 (1, n_feature)으로 표현가능하며 하나의 단백질은 L개의 아미노산을 갖기 때문에 (L, n_feature)으로 표현이 가능합니다.
2D 데이터는 아미노산(residue) 간의 관계를 표현하고 있기 때문에 (L, L)로 표현됩니다.
이를 하나의 데이터로 합치기 위해서 i, j 픽셀에 i 1D 데이터와 j 1D 데이터를 연결하여 뒤에 병합하게 되면 (L, L, 2 * n_feat + 1)로 구성되게 됩니다.
2.2 Method
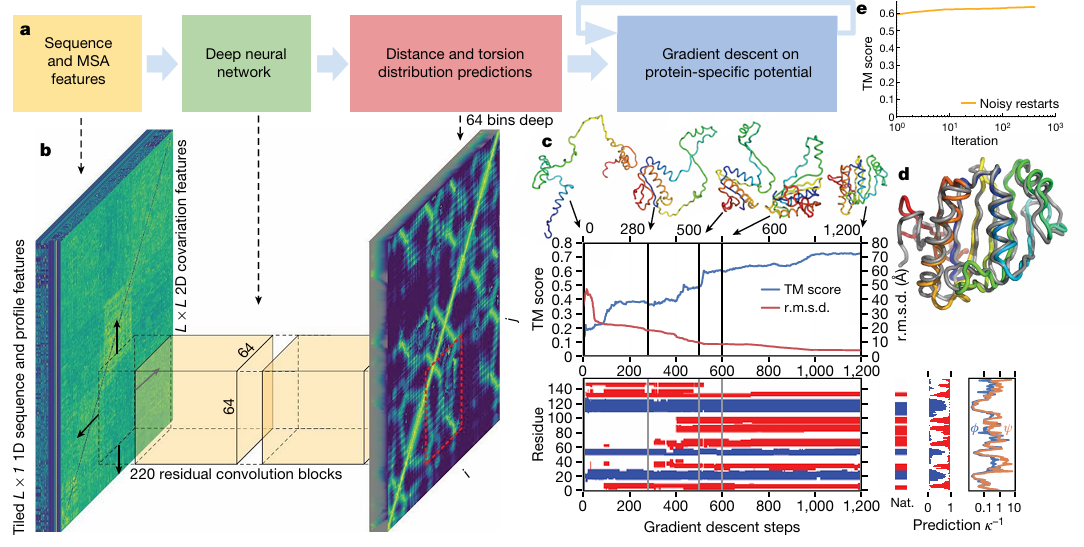
CASP13 타겟 T0986s2 단백질을 대상으로 단백질 구조 예측 과정을 시각적으로 나타내고 있습니다.
AlphaFold 1의 구조는 다음과 같은 단계로 구성됩니다:
- MSA 기반 특징 추출
- 220개의 residual block으로 구성된 Dilated Convolutional Residual Network를 통한 거리 예측
- Gradient descent를 통한 3D 구조 최적화
2.2.1 Distogram Prediction
LxL distogram은 단백질의 L개의 residue 쌍 사이의 거리 분포를 나타내는 행렬입니다. 이 행렬은 다음과 같은 특징을 가집니다:
- 각 셀 (i, j)는 residue i, j 간 거리 dij에 대한 확률 분포를 표현합니다. 이러한 확률 분포는 구조 예측의 불확실성을 반영합니다.
- 거리 범위(2Å~22Å)를 64개의 bin으로 나누어 예측합니다. 이러한 binning은 거리 분포를 더 세밀하게 모델링할 수 있게 합니다.
- 확률적 표현을 통해 구조 예측의 불확실성을 반영합니다. 이러한 불확실성은 구조 예측 모델의 출력에 대한 신뢰도를 평가하는 데 사용됩니다.
주요 특징:
- 64x64 크기의 블록으로 분할하여 학습
- Add noise (과적합 방지를 위한 전략) 적용
- 전체 단백질 체인에 대해 sliding window 방식으로 distogram 계산
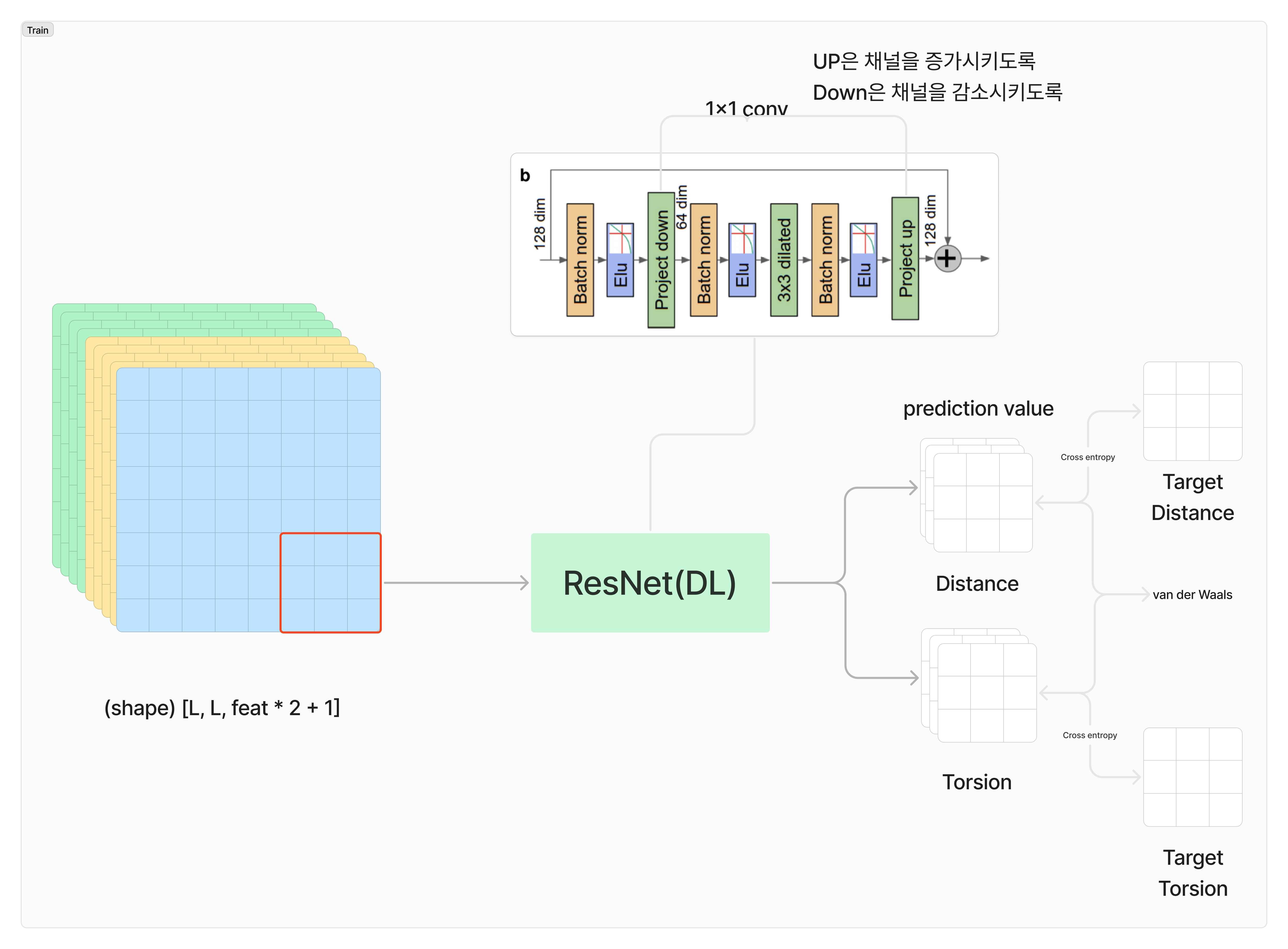
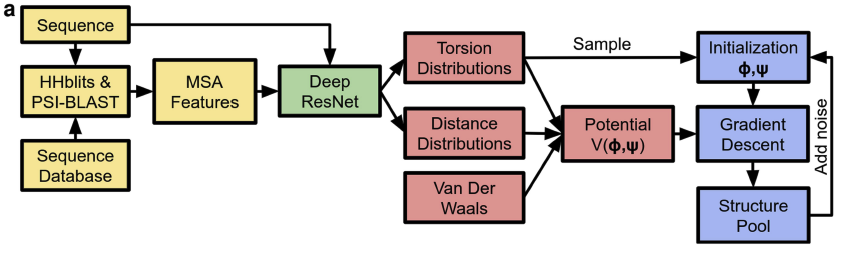
2.2.2 Neural Network Architecture
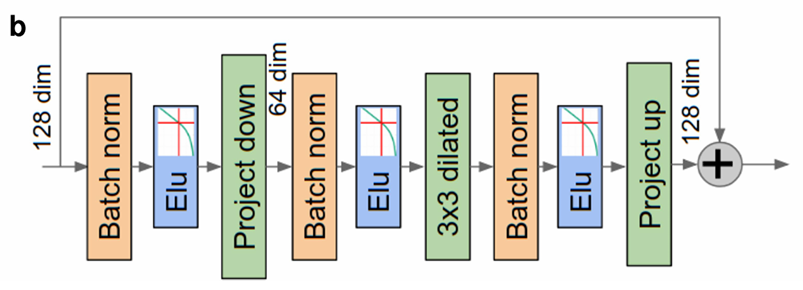
AlphaFold 1의 신경망 구조는 일반적인 CNN과 다른 특별한 설계 원칙을 따릅니다:
- Pooling Layer 미사용 이유:
- 단백질 구조 예측에서는 입력과 출력의 공간적 해상도가 동일해야 함()
- Pooling은 공간 정보를 손실시켜 residue 간 정확한 거리 예측을 방해할 수 있음
- 모든 residue 쌍 간의 관계를 보존하기 위해 downsampling을 피함
- Dilated Convolution 사용 이유:
- 넓은 수용영역(receptive field)을 확보하면서도 파라미터 수를 효율적으로 유지
- 멀리 떨어진 residue 간의 장거리 상호작용을 포착 가능
- Pooling 없이도 전역적 문맥 정보를 획득 가능
- Input-Output Size 유지 필요성:
- 각 residue 쌍에 대한 거리 분포를 예측하기 위해 LxL 크기 유지 필요
- 모든 residue 쌍의 관계를 누락없이 표현하기 위함
- Distogram 예측을 위해 공간적 해상도 보존이 중요
주요 구성 요소:
- 220개의 residual block으로 구성된 깊은 네트워크
- Skip connection을 통한 gradient vanishing 문제 해결
- ELU 활성화 함수로 비선형성 확보
- Batch normalization과 dropout으로 정규화
- 최종 출력은 각 residue 쌍의 거리를 64개 bin으로 예측
2.2.3 Structure Generation
AlphaFold 1의 구조 생성 과정은 다음과 같은 단계로 이루어집니다:
- 입력 데이터 처리
- 단백질 서열과 MSA(Multiple Sequence Alignment) 정보를 입력으로 받음
- 1D, 2D 특징을 추출하여 신경망의 입력으로 변환
- 신경망을 통한 예측
- Deep Neural Network가 거리와 torsion angle 분포를 예측
- 64개 bin으로 나눈 distogram 형태로 residue 간 거리 예측
- 각 residue의 torsion angle (φ, ψ) 분포 예측
- 구조 최적화
- 예측된 거리와 각도 분포를 바탕으로 potential 함수 구성
- Gradient descent를 통해 potential을 최소화하는 3D 구조 탐색
- 물리적 제약조건과 Van der Waals 힘을 고려한 구조 정제
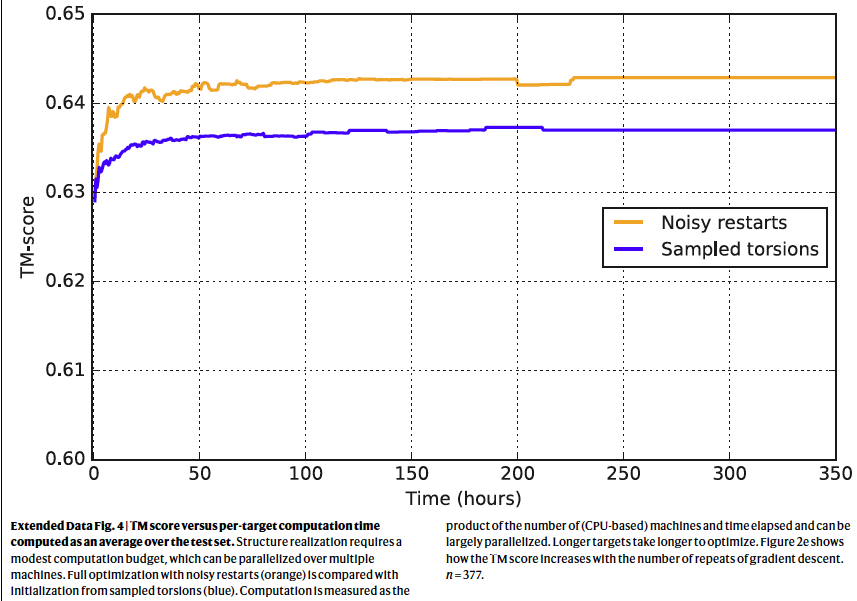
- 여러 번의 noisy restart를 통해 다양한 구조 샘플링
Torsion angle 초기화의 중요성:
- 신경망은 각 residue의 torsion angle에 대해 discrete 확률 분포를 예측 (10° 간격, 1,296 bins)
- 이 분포에서 sampling하여 초기 구조를 생성하는데, 이는 단백질의 이차 구조(secondary structure)를 부분적으로 결정
- 예측된 분포가 multimodal일 수 있어, von Mises 분포로 fitting하여 단일 모드로 통합
- 이러한 초기화는 gradient descent가 더 나은 local optimum을 찾도록 도움
학습 과정:
- MSA 기반 특징과 실제 구조 간의 관계를 end-to-end로 학습
- Cross-entropy loss를 사용하여 거리와 각도 분포 예측을 최적화
- 보조 테스크(이차 구조, 표면 접근성 예측 등)를 통한 학습 보조
- 27개의 CASP11 FM 도메인으로 검증하며 early stopping 적용
3. 종합(시사점, 한계점)
-
@이호민
그간 의학과 생물학 분야의 어려운 용어들 때문에 쉽게 접근하기 힘들었지만, 제가 조금이나마 알고 있는 딥러닝을 접목한 AlphaFold 논문 덕분에 리뷰를 진행할 수 있었습니다. 특히 약학을 전공하신 @기백 김 님의 배경지식 공유는 가뭄의 단비와 같았습니다. 🙇 또한 단백질의 기능이 3차원 구조로부터 발현된다는 점이 매우 흥미롭고 신기했습니다.
이 논문의 특징은 새로운 구조를 제시하기보다는 특정 문제를 해결하기 위한 구체적인 솔루션을 제시한다는 점입니다. 기존에 제가 읽었던 논문들이 새로운 모델이나 optimizer 등 방법론 중심이었던 반면, 이 논문은 문제 해결을 위한 방법들을 논리적으로 서술하는 데 중점을 두고 있습니다. 오히려 딥러닝 논문이라기보다는 Kaggle 솔루션에 더 가까운 느낌이었습니다.
논문에서 특히 감명 깊었던 세 가지 부분은
#distogram,#potential 함수와 loss function의 관계,#AlphaFold에서의 CNN입니다.-
#Distogram&#potential 함수와 loss 함수의 관계논문에서는 거리와 residue 간의 각도를 regression 문제가 아닌 classification 문제로 해석한 점이 흥미롭다(2.2.1 Distogram Prediction에 설명되어 있음). 그 이유를 추정해보면, 본 문제에서 다루는 distance와 torsion 값들은 binning 처리가 적합하다고 해석할 수 있다. distance를 binning하지 않고 MSE와 같은 방법으로 loss function을 정의했다면, 대부분의 distance가 큰 값(22 또는 22 이상)을 가졌을 것이다. 이는 학습하기 어려웠을 것이며, 상대적으로 더 중요하고 어려운 작은 distance 값들의 예측이 힘들었을 것이다. residue 간의 거리를 확률 분포로 예측함으로써 정확도뿐만 아니라 예측의 불확실성도 추정할 수 있다는 점이 주목할 만하다.
확률 분포로 근사하는 방법의 적합성은 여기서 그치지 않는다. 이는 단백질이 접혀가는 과정에서 사용되어 온 potential 함수를 딥러닝으로 해석하기 위한 적절한 방법으로 보인다. 논문의 그림에서 보듯이, 처음에 긴 구조였던 단백질이 학습을 통해 점차 접혀가는 모습이 이를 잘 보여준다.
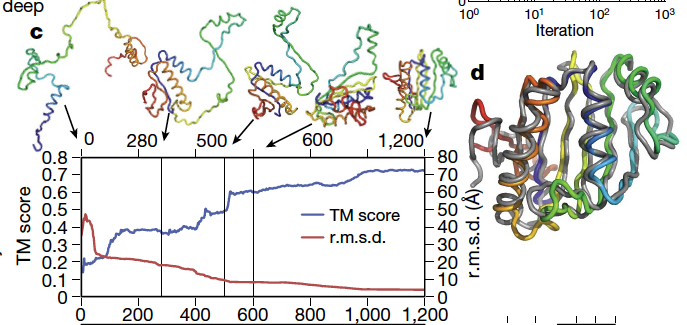
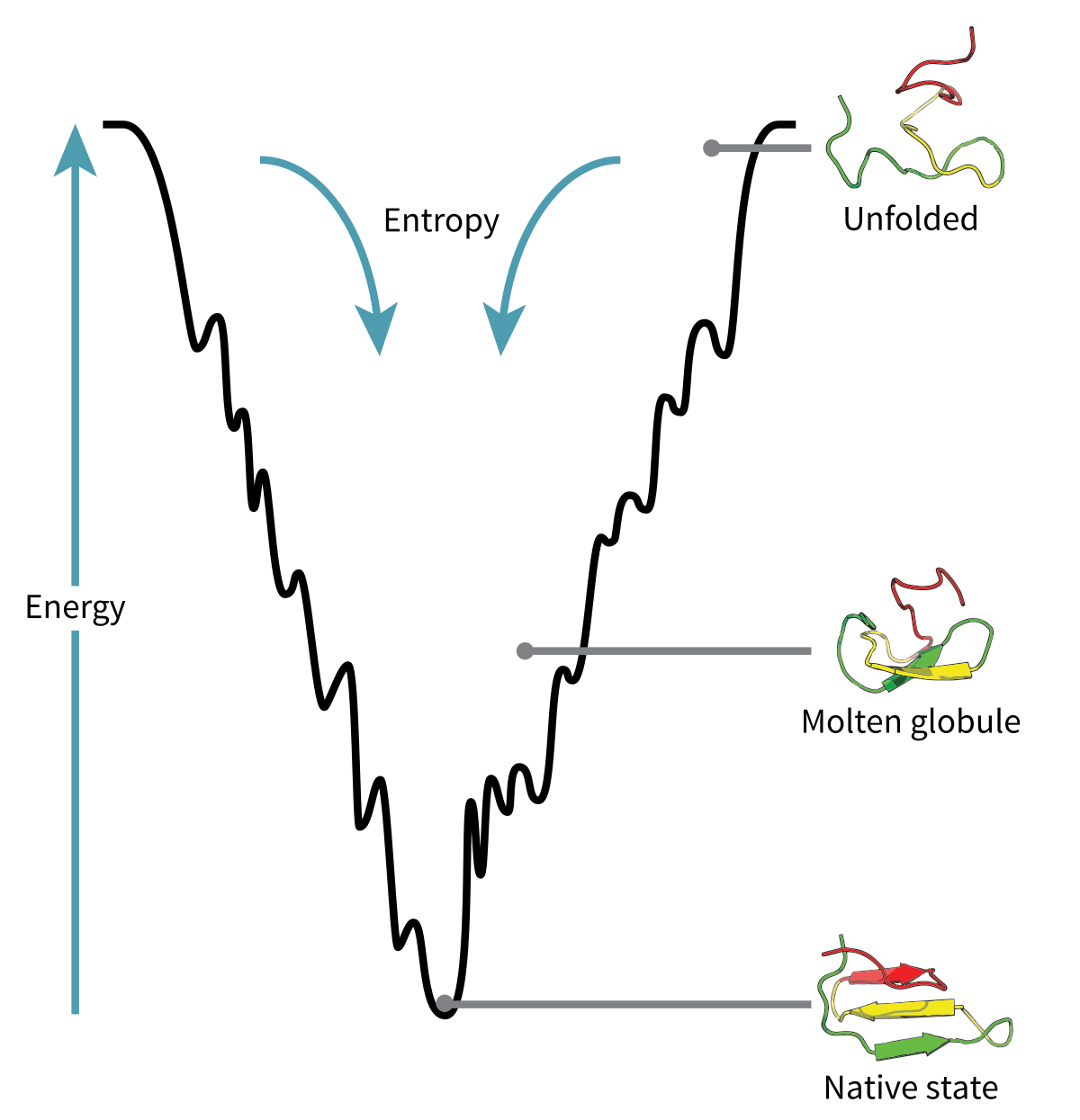
-
#AlphaFold에서의 CNNAlphaFold에서는 일반적으로 사용되는 CNN과 다른 특징이 있는데, 바로 풀링 레이어를 사용하지 않고 확장 합성곱(dilation convolution)을 사용한다는 점이다. 일반적인 CNN에서 풀링 레이어를 사용하는 이유는 이미지 처리 시 주변 픽셀 정보를 종합하여 정보를 이해하는 것이 중요하기 때문이다. 하지만 AlphaFold가 해결하고자 하는 문제는 이와는 다르다. AlphaFold에서 하나의 픽셀은 두 residue 간의 거리를 의미하며, 대각행렬 인근의 픽셀들은 서열상 가까운 residue를 나타낸다. 따라서 인근 픽셀의 정보보다는 서열상 멀리 떨어진 residue 간의 관계를 파악하는 것이 더 중요하다. 이러한 이유로 풀링 레이어 대신 확장 합성곱 레이어를 채택했다고 볼 수 있다. 또한 64×64 크기의 입력을 동일한 크기로 예측해야 하는데, 풀링 레이어로 인해 줄어든 특징 맵(feature map)의 크기를 다시 늘리는 것이 기술적으로 어렵다는 문제도 있다.
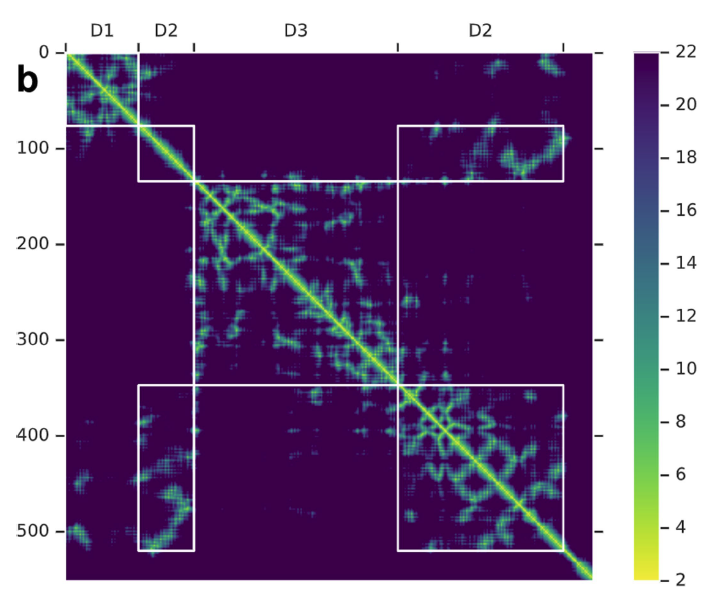
-
-
@세영 최
AlphaFold 논문을 리뷰하면서 현실 세계의 문제를 해결하기 위해 딥러닝을 어떻게 적용해야 하는지 조금이나마 통찰력을 얻을 수 있었습니다. 기존에 공부했던 딥러닝을 적용하는 문제들은 대부분 데이터로부터 정보를 얻거나 패턴을 분석하는 데 초점이 맞춰져 있었습니다. 그렇기 때문에 어떻게 딥러닝을 사용해서 복잡한 물리적 현상을 모델링하고 예측하는 지 항상 궁금했었는데, 이런 부분이 해소가 된 것 같습니다.
논문에서 가장 흥미롭게 본 부분은 AlphaFold의 메인 아이디어 중 하나인 LxL distogram과 Dilated Convolution 입니다.
LxL distogram은 각각의 셀이 단백질의 두 residue 간의 거리에 대한 확률 분포를 나타내는데, 이를 64개의 bin으로 쪼개 연속적인 dij 값을 이산적으로 표현하게 됩니다. (2.2.1 Distogram Prediction 참고)
덕분에 모델이 특정한 dij를 예측하는 것이 아닌 확률 분포로 나타냄으로써 예측의 불확실성을 표현할 수 있습니다. 예측이 명확한 경우에는 분포가 특정 bin에 집중되고, 불확실한 경우 여러 bin에 걸쳐서 분포가 확산됩니다.
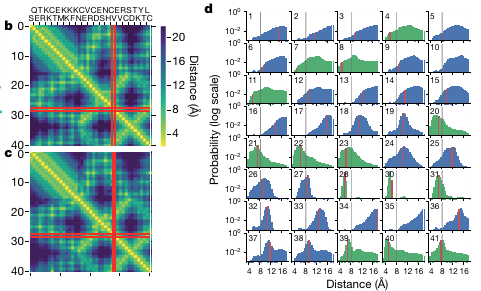
AlphaFold는 distance와 tortion, Vscore2_smooth (Van Der Waals)를 사용해 Potential function을 정의하고, Gradient descent 알고리즘을 통해 최적화하게 됩니다.
두 번째로, @2.2.2 Neural Network Architecture의 Dilated Convolution 부분이 개인적으로 흥미로웠습니다. 해당 절에서 언급된 것처럼 일반적인 CNN에서는 사용하지 않는 Dilated Convolution을 채택했으며, 또한 풀링 레이어도 사용하고 있지 않습니다. 또한 활성화 함수로도 생소한 ELU를 사용하고 있다는 점이 흥미롭습니다.
Dilated Convolution을 사용하면 특정 위치의 정보가 누락될 수 있지만, 전역 정보를 학습할 수 있다는 장점이 있습니다. 또한 풀링 레이어를 생략하여 residue 쌍의 정확한 상호작용 정보를 남겨 고해상도 예측을 가능하게 합니다.
출력 편향 문제 해결
Elu 활성화 함수의 경우에는 일반적으로 사용하는 relu에 비해 음의 영역에서도 0이 아닌 음수 값을 가지게 됩니다. 덕분에 양수, 음수 값을 모두 포함하게 되어 출력 분포가 대칭적으로 나타나게 합니다. AlphaFold에서는 Elu, Batch norm을 사용하여 출력 평균이 0에서 멀어지는 문제를 해결하고 있습니다.
이번 AlphaFold 논문을 읽으면서 생물학 기본 지식이 부족했던 탓인지 논문을 이해하는 데 많은 어려움을 겪었습니다. 하지만 @기백 김님께서 배경 지식을 이해하는 데 큰 도움을 주셨고, @이호민님께서 이해가 부족했던 부분을 명확히 설명해주신 덕분에 논문을 제대로 이해할 수 있었습니다. 두 분께 진심으로 감사드립니다.
-
@기백 김
- 시사점
1) 생물학적 연구와 응용의 확대 가능성
AlphaFold는 단백질-단백질 상호작용, 신약 개발, 질병 메커니즘 이해 등 다양한 생물학적 문제에서 활용될 수 있는 강력한 도구입니다. 특히, 실험적으로 구조가 밝혀지지 않은 단백질의 연구를 가속화할 수 있습니다.
2) 딥러닝의 물리적 현상 모델링 가능성
딥러닝을 통해 단백질의 접힘 과정을 모델링함으로써 복잡한 물리적 현상을 예측하는 새로운 가능성을 열었습니다. 이는 생물학 외에도 화학, 물리학 등 다양한 과학 분야에 응용될 잠재력이 있습니다.
3) 분야 간 협력의 중요성
AlphaFold 연구는 데이터 과학과 생물학이라는 서로 다른 두 분야의 협력이 얼마나 중요한 성과를 이끌어낼 수 있는지를 보여주는 사례입니다. 딥러닝 알고리즘과 생물학적 지식이 조화를 이루었을 때의 강점을 잘 증명합니다.
- 한계점
1) 데이터 의존성
AlphaFold는 다중 서열 정렬(MSA)의 품질에 크게 의존하며, 상동 서열이 적은 경우 성능 저하가 발생할 가능성이 있습니다. 희귀 단백질 서열 데이터에 대한 적용성을 개선하는 연구가 필요합니다.
2) 구조 복잡성에서의 제한
AlphaFold는 단순한 도메인이나 비교적 짧은 단백질 구조에서 탁월한 성능을 발휘했지만, 복잡하고 긴 단백질 구조의 예측에서는 여전히 개선의 여지가 있습니다.
3) 실험적 데이터와의 차이
AlphaFold의 예측 구조는 많은 경우 실험적으로 검증된 구조와 유사하지만, 특정한 세부적인 차이가 존재합니다. 이는 예측 퍼텐셜 설정 및 최적화 과정의 개선이 필요한 부분입니다.
- 가장 흥미로웠던 부분
1) 퍼텐셜 함수의 설계
AlphaFold의 퍼텐셜 함수 설계는 단백질 구조 예측의 성공에서 핵심적인 역할을 합니다. 이 함수는 단백질 구조를 나타내는 여러 물리적 제약과 예측된 정보를 통합하여, 에너지 공간에서 가장 안정적인 구조를 찾도록 설계되었습니다. 퍼텐셜 함수는 크게 세 가지 주요 요소로 구성됩니다: 잔기 간 거리 기반 퍼텐셜(Vdistance), 토션 각도 기반 퍼텐셜(Vtorsion), 그리고 스테릭 충돌 방지 항(Vscore2_smooth)입니다.
- 거리 기반 퍼텐셜 (Vdistance)
AlphaFold는 잔기 간 거리 정보를 distogram 형식으로 예측하여 이를 기반으로 퍼텐셜을 계산합니다. 잔기 i와 j 사이의 거리 dij를 확률 분포
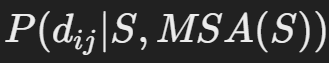
로 표현하며, 이를 음의 로그 확률 형태로 변환하여 에너지를 계산합니다.
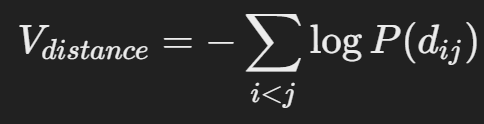
이 퍼텐셜은 단백질의 전역적 기하학적 특성을 반영하며, 잔기 간의 물리적 배치를 결정하는 데 중요한 역할을 합니다.
- 토션 각도 기반 퍼텐셜 (Vtorsion)
단백질 구조에서 backbone 회전각(Φ, Ψ)은 구조의 형태와 안정성을 결정짓는 중요한 변수입니다. AlphaFold는 각 잔기의 Φ와 Ψ 값을 확률 분포
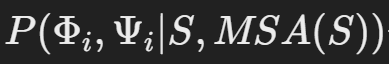
로 예측하며, 이를 기반으로 부드러운 퍼텐셜 항을 생성합니다.
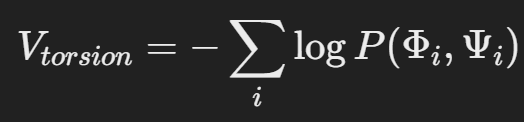
예측된 Φ, Ψ 값은 단백질의 2차 구조(예: α-나선, β-병풍)를 결정하며, 잔기 간 거리를 최적화하기 위한 주요 기준점이 됩니다. 이 과정에서 Φ와 Ψ 값이 결정되면, 잔기 간 거리와 공간적 배치가 결정되며 퍼텐셜 값 또한 정해집니다. 이는 단백질 접힘 과정을 효율적으로 모델링하는 데 필수적인 요소입니다.
- 스테릭 충돌 방지 퍼텐셜 (Vscore2_smooth)
단백질의 구조 내에서 물리적 충돌(스테릭 충돌)을 방지하기 위해 Rosetta의 Vscore2_smooth 항을 사용합니다. 이 항목은 반데르발스 힘과 같은 물리적 제약을 포함하며, 구조의 현실성을 보장하고 비합리적인 구조 형성을 방지합니다.
- 참조 분포와 보정
AlphaFold는 거리와 각도 퍼텐셜이 과도하게 특정 값에 치우치지 않도록, 참조 분포를 사용하여 일반화된 에너지 공간을 형성합니다. 참조 분포는 단백질 서열과 독립적으로 평균적인 거리 분포를 모델링하며, 이를 통해 서열 특성에 의존하지 않는 구조 예측이 가능해집니다.
- 최종 퍼텐셜 함수
AlphaFold의 최종 퍼텐셜 함수는 다음과 같이 표현됩니다.

여기서 각 항목은 가중치 λ를 통해 조정되며, 기본적으로 가중치는 균등하게 설정됩니다. 퍼텐셜 함수는 AlphaFold의 예측 과정에서 신경망이 출력하는 확률적 정보를 물리적 구조로 변환하는 데 핵심적인 역할을 합니다. 특히 Φ와 Ψ 각도가 결정되면 퍼텐셜 값이 정해지며, 이를 기반으로 Gradient Descent 알고리즘이 작동하여 구조를 최적화합니다.
2) Gradient Descent Algorithm을 이용한 최적화 과정
AlphaFold는 퍼텐셜 함수를 최소화하여 단백질의 3차원 구조를 완성합니다. 초기 구조는 예측된 distogram과 Φ, Ψ 각도를 기반으로 설정되며, Gradient Descent Algorithm을 통해 점진적으로 개선됩니다.
- 최적화 과정의 주요 단계
초기 구조는 예측된 토션 각도 분포에서 샘플링된 값을 사용하여 생성됩니다. 각 Gradient Descent 단계에서 퍼텐셜 함수의 기울기를 계산하여, 가장 낮은 에너지 상태로 구조를 이동시킵니다. 과정이 반복될수록 구조는 점차 접히며, 잘못된 접힘이 수정되고 최적의 상태에 도달합니다.
- Noisy Restarts
AlphaFold는 초기화 단계에서 작은 노이즈를 추가하여 여러 구조를 생성하고, 이들 중 가장 낮은 에너지를 가진 구조를 선택합니다. 이 접근법은 지역 최적점에 갇히는 문제를 방지하고, 더 다양한 구조를 탐색할 수 있도록 합니다.
- 최적화의 수렴
몇백 번의 Gradient Descent 반복 후, 구조는 퍼텐셜 함수의 지역 최적점에 도달하며 TM 점수가 꾸준히 향상됩니다.
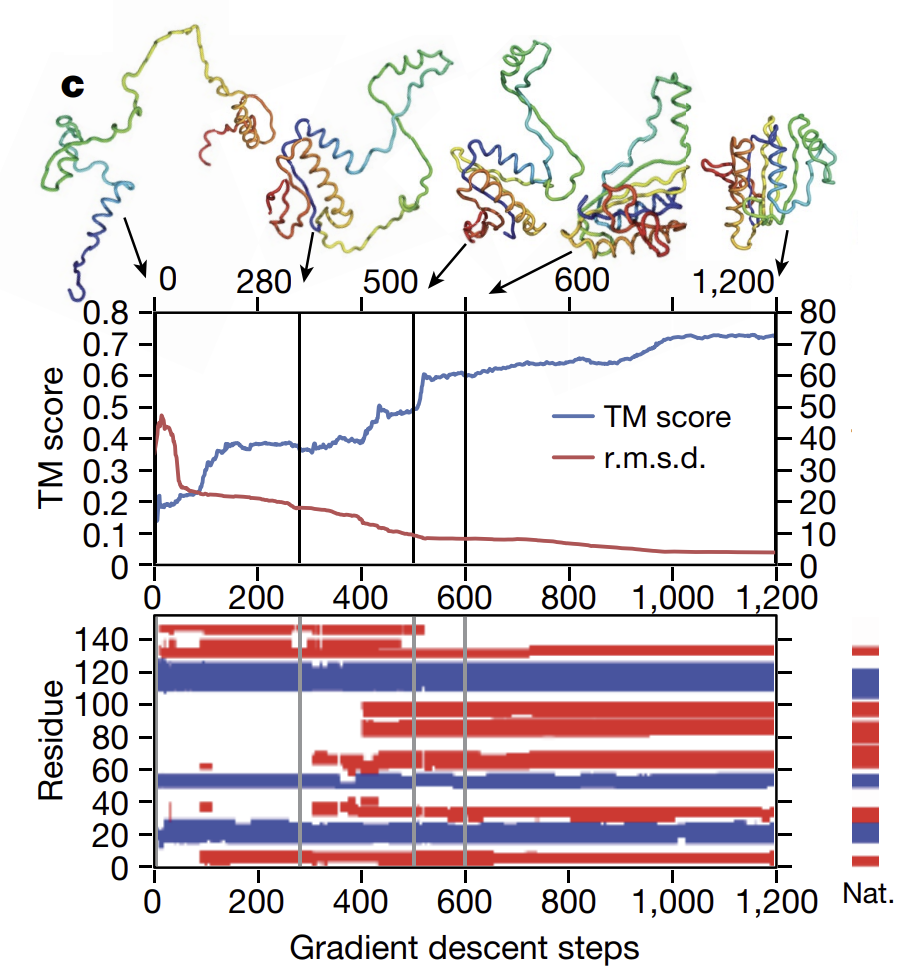
는 최적화 과정에서 TM 점수가 증가하는 모습을 시각적으로 보여줍니다. Gradient Descent Algorithm을 활용한 최적화는 AlphaFold가 기존 방법에 비해 훨씬 더 효율적이고 정확한 구조 예측을 가능하게 한 핵심 요소입니다. 이 과정은 단백질의 접힘을 물리적 에너지 공간에서 모델링하며, 전례 없는 정확도를 달성할 수 있도록 했습니다.
- 느낀 점
약학(Pharmacy)을 전공하는 입장에서, 이 연구는 단백질 구조 예측이 약물 설계와 생물학적 기능 분석에서 얼마나 중요한 역할을 하는지 다시금 깨닫게 했습니다. 특히 단백질의 3차원 구조가 Φ, Ψ 두 개의 각도로 결정될 수 있다는 점이 매우 인상적이었습니다. 이렇게 단순한 변수들로 복잡한 생물학적 구조를 설명할 수 있다는 점은 놀라웠으며, 딥러닝 기술이 이를 구현한 방식에서 큰 경이로움을 느꼈습니다.
또한, 데이터 과학(Data Science) 전공자들과 함께 논문을 읽고 토론하며, 신경망 설계 원리와 딥러닝 기술이 생물학적 문제 해결에 어떻게 적용되는지 깊이 이해할 수 있었습니다. Dilated Convolution을 통한 전역적 관계 학습이나 그래디언트 디센트를 활용한 최적화 과정은, 생물학적 배경만으로는 알기 어려웠던 새로운 통찰을 제공했습니다.
결론적으로, AlphaFold 연구는 약학과 데이터 과학이 협력할 때 얼마나 강력한 시너지를 낼 수 있는지를 보여주는 훌륭한 사례였습니다. 앞으로 이러한 기술이 더 발전하여 약물 설계와 단백질 기능 연구를 혁신적으로 변화시킬 것을 기대합니다.
Reference
[1]AlphaFold 1 설명
| [AlphaFold: Improved protein structure prediction […] | AI & Molecular World | Andrew Senior](https://www.youtube.com/watch?v=HRqf76ULlCU) |
[2]AlphaFold code
deepmind-research/alphafold_casp13 at master · google-deepmind/deepmind-research
[3] How to work dilated convolution layer